pet-surface - Surface-based processing of PET images¶
This pipeline performs several processing steps for the analysis of PET data on the cortical surface [Marcoux et al., 2018]:
- co-registration of PET and T1-weighted MR images;
- intensity normalization;
- partial volume correction;
- robust projection of the PET signal onto the subject’s cortical surface;
- spatial normalization to a template;
- atlas statistics.
This pipeline relies mainly on tools from FreeSurfer and PETPVC [Thomas et al., 2016].
Clinica & BIDS specifications for PET modality
Since Clinica v0.6, PET data following the official specifications in BIDS version 1.6.0 are now compatible with Clinica.
See BIDS page for more information.
Prerequisite¶
You need to have performed the t1-freesurfer pipeline on your T1-weighted MR images.
Dependencies¶
If you only installed the core of Clinica, this pipeline needs the installation of FreeSurfer 6.0, FSL 6.0, and PETPVC 1.2.4 (which depends on ITK 4) on your computer.
In addition, you also need to either install SPM12 and Matlab, or spm standalone.
Running the pipeline¶
The pipeline can be run with the following command line:
clinica run pet-surface [OPTIONS] BIDS_DIRECTORY CAPS_DIRECTORY ACQ_LABEL
{pons|cerebellumPons|pons2|cerebellumPons2} PVC_PSF_TSV
where:
BIDS_DIRECTORYis the input folder containing the dataset in a BIDS hierarchyCAPS_DIRECTORYis the output folder containing the results in a CAPS hierarchyACQ_LABELis the label given to the PET acquisition, specifying the tracer used (trc-<acq_label>).- The reference region is used to perform intensity normalization (i.e. dividing each voxel of the image by the average uptake in this region) resulting in a standardized uptake value ratio (SUVR) map.
It can be
cerebellumPonsorcerebellumPons2(used for amyloid tracers) andponsorpons2(used for FDG). See PET introduction for more details about masks versions. -
PVC_PSF_TSVis the TSV file containing thepsf_x,psf_yandpsf_zof the PSF for each PET image. It is expected to be generated by the user. More explanation is given in PET Introduction page.Clinica v0.3.8+
Since the release of Clinica v0.3.8, the handling of PSF information has changed. In previous versions of Clinica, each BIDS-PET image had to contain a JSON file with the
EffectiveResolutionInPlaneandEffectiveResolutionAxialfields corresponding to the PSF in mm.EffectiveResolutionInPlaneis replaced by bothpsf_xandpsf_y,EffectiveResolutionAxialis replaced bypsf_zand theacq_labelcolumn has been added. Additionally, the SUVR reference region is now a compulsory argument: it will be easier for you to modify Clinica if you want to add a custom reference region (PET Introduction page). ChoosecerebellumPonsfor amyloid tracers orponsfor FDG to have the previous behavior.
with specific options :
-rec/--reconstruction_method: Select only images based on a specific reconstruction method.
Optional parameters common to all pipelines
-tsv/--subjects_sessions_tsv
This flag allows you to specify in a TSV file the participants belonging to your subset. For instance, running the FreeSurfer pipeline on T1w MRI can be done using :
clinica run t1-freesurfer BIDS_PATH OUTPUT_PATH -tsv my_subjects.tsv
participant_id session_id
sub-CLNC0001 ses-M000
sub-CLNC0001 ses-M018
sub-CLNC0002 ses-M000
Creating the TSV
To make the display clearer the rows here contain successive tabs but that should not happen in an actual TSV.
-wd/--working_directory
By default when running a pipeline, a temporary working directory is created. This directory stores all the intermediary inputs and outputs of the different steps of the pipeline. If everything goes well, the output directory is eventually created and the working directory is deleted.
With this option, a working directory of your choice can be specified. It is very useful for the debugging process or if your pipeline crashes. Then, you can relaunch it with the exact same parameters which will allow you to continue from the last successfully executed node.
For the pipelines that generate many files, such as dwi-preprocessing (especially if you run it on multiple subjects), a specific drive/partition with enough space can be used to store the working directory.
-np/--n_procs
This flag allows you to exploit several cores of your machine to run pipelines in parallel, which is very useful when dealing with numerous subjects and multiple sessions.
Thanks to Nipype, even for a single subject, a pipeline can be run in parallel by exploiting the cores available to process simultaneously independent sub-parts. We recommend using your_number_of_cpu - 1 for costly pipelines such as pet-surface-longitudinal.
If you do not specify -np / --n_procs flag, Clinica will detect the number of threads to run in parallel and propose the adequate number of threads to the user.
-cn/--caps-name
Use this option if you want to specify the name of the CAPS dataset that will be used inside the dataset_description.json file, at the root of the CAPS folder (see CAPS Specifications for more details). This works if this CAPS dataset does not exist yet, otherwise the existing name will be kept.
 Please note that PETPVC is extremely demanding in terms of resources and
may cause the pipeline to crash if many subjects happen to be partial volume corrected at the same time
(
Please note that PETPVC is extremely demanding in terms of resources and
may cause the pipeline to crash if many subjects happen to be partial volume corrected at the same time
(Error : Failed to allocate memory for image).
To mitigate this issue, you can do the following:
- Use a working directory when you launch Clinica (see option
--working-directory) - If the pipeline crashes, just re-launch the command with the same working directory. The whole processing will continue where it left. You can reduce the number of threads to run in parallel the second time.
Tip
Do not hesitate to type clinica run pet-surface --help to see the full list of parameters.
Known error on macOS with gtmseg
If you are running pet-surface or pet-surface-longitudinal on macOS, we noticed that if the path to the CAPS is too long, the pipeline fails when the gtmseg command from FreeSurfer is executed.
This generates crash files with gtmseg in the filename, for instance:
$ nipypecli crash crash-20210404-115414-sheldon.cooper-gtmseg-278e3a57-294f-4121-8a46-9975801f24aa.pklz
[...]
Abort
ERROR: mri_gtmseg --s sub-ADNI011S4105_ses-M000 --usf 2 --o gtmseg.mgz --apas apas+head.mgz --no-subseg-wm --no-keep-cc --no-keep-hypo
gtmseg exited with errors
Standard error:
Saving result to '<caps>/subjects/sub-ADNI011S4105/ses-M000/t1/freesurfer_cross_sectional/sub-ADNI011S4105_ses-M000/tmp/tmpdir.xcerebralseg.50819/tmpdir.fscalc.53505/tmp.mgh' (type = MGH ) [ ok ]
Saving result to '<caps>/subjects/sub-ADNI011S4105/ses-M000/t1/freesurfer_cross_sectional/sub-ADNI011S4105_ses-M00/tmp/tmpdir.xcerebralseg.50819/tmpdir.fscalc.53727/tmp.mgh' (type = MGH ) [ ok ]
Saving result to '<caps>/subjects/sub-ADNI011S4105/ses-M000/t1/freesurfer_cross_sectional/sub-ADNI011S4105_ses-M000/tmp/tmpdir.xcerebralseg.50819/tmpdir.fscalc.53946/tmp.mgh' (type = MGH ) [ ok ]
Return code: 1
This is under investigation (see Issue #119 for details) and will be solved as soon as possible.
Outputs¶
Results are stored in the following folder of the
CAPS hierarchy:
subjects/<participant_id>/<session_id>/pet/surface
The main output files are (where * stands for <participant_id>_<session_id>):
*_trc-<label>_pet_space-<label>_suvr-<label>_pvc-iy_hemi-<label>_fwhm-<value>_projection.mgh: PET data that can be mapped onto meshes. If thespaceisfsaverage, it can be mapped either onto the white or pial surface of FsAverage. If thespaceisnative, it can be mapped onto the white or pial surface of the subject’s surface (i.e.{l|r}h.white,{l|r}h.pialfiles from thet1-freesurferpipeline).*_hemi-{left|right}_midcorticalsurface: surface at equal distance between the white matter/gray matter interface and the pial surface (one per hemisphere).atlas_statistics/*_trc-<label>_pet_space-<label>_pvc-iy_suvr-<label>_statistics.tsv: TSV files summarizing the regional statistics on the labelled atlases (Desikan and Destrieux).
Note
The full list of output files from the pet-surface pipeline can be found in The ClinicA Processed Structure (CAPS) specifications.
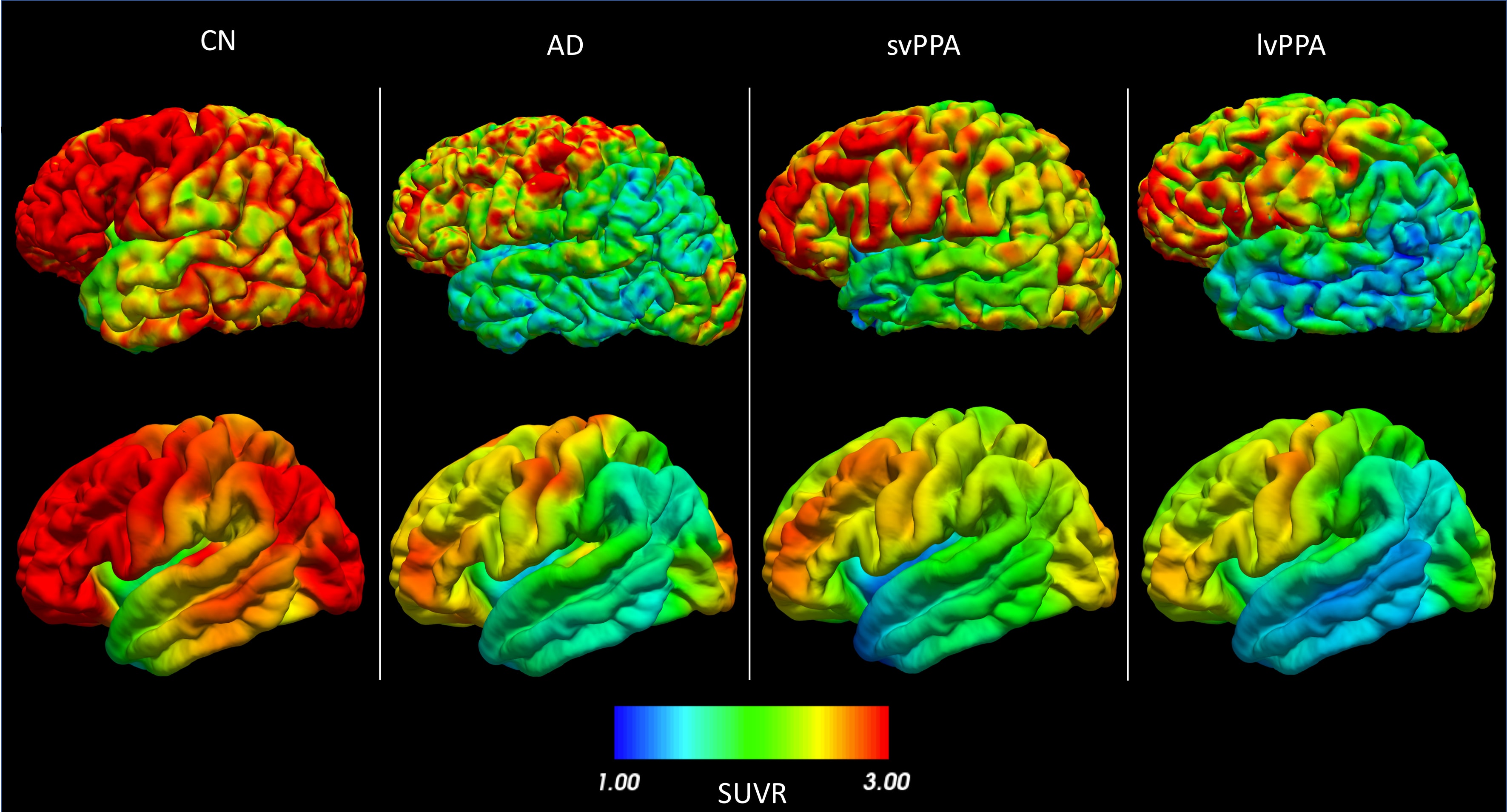
Going further¶
- You can use projected PET data to perform group comparison or correlation analysis with the
statistics-surfacepipeline. - You can use projected PET data to perform classification based on machine learning, as showcased in the AD-ML framework.
Describing this pipeline in your paper¶
Example of paragraph:
These results have been obtained using the pet-surface pipeline of Clinica
[Routier et al., 2021;
Marcoux et al., 2018].
The subject’s PET image was registered to the T1-weighted MRI using spmregister
(FreeSurfer) and intensity normalized using
the [pons | pons and cerebellum] from the Pick atlas in MNI space as reference region
(registration to MNI space was performed using
SPM12).
Partial volume correction was then performed using the iterative Yang algorithm implemented in
PETPVC
[Thomas et al., 2016]
with regions obtained from gtmseg (FreeSurfer).
Based on the subject’s white surface and cortical thickness,
seven surfaces for each hemisphere were computed,
ranging from 35% to 65% of the gray matter thickness.
The partial volume corrected data were projected onto these meshes and
the seven values were averaged, giving more weight to the vertices near the center of the cortex.
Finally, the projected PET signal in the subject’s native space was
spatially normalized to the standard space of FsAverage
(FreeSurfer).
Tip
Easily access the papers cited on this page on Zotero.
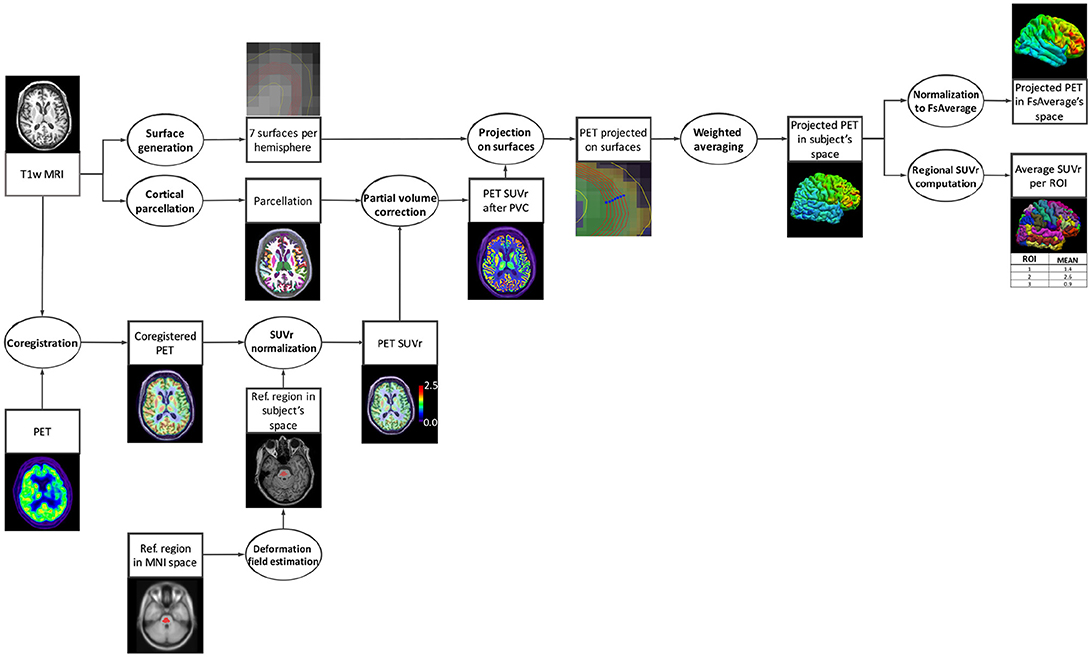
Appendix I: Diagram of the pipeline execution¶
Appendix II: How to manipulate outputs¶
Outputs of pipeline are composed of two different types of file: surface files and MGH data that are to be overlaid onto a surface.
Surface file¶
Surface files can be read using various software packages.
You can open them using freeview (FreeSurfer viewer), with freeview -f /path/to/your/surface/file.
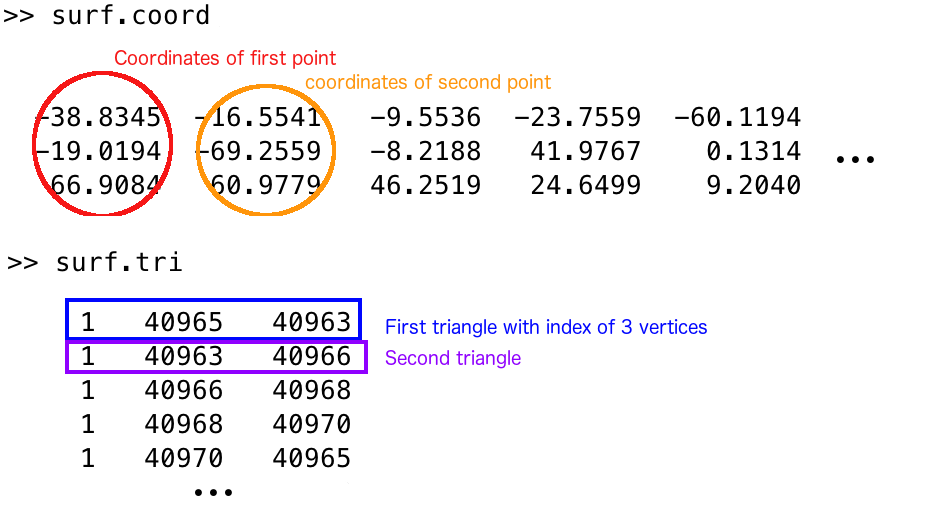
You can also open them in MATLAB, using SurfStat: mysurface = SurfStatReadSurf('/path/to/your/surface/file').
This will give you a structure with fields coord (for coordinates), a list of coordinates for each point of the mesh, and also the field tri (for triangle), a list of triplet for each triangle, each number representing the Nth vertex of the coord list.
Below is an example to make things clearer (read with Matlab).
Data files¶
Data files wear the .mgh extension.
They are composed of a single vector.
These files contain a vector, where the value at the Nth position must be mapped into the Nth vertex of the coord list to be correctly represented.
You can access them either in Matlab with the command:
mydata = SurfStatReadData('/path/to/your/file.mgh');
(you will get a single row vector),
or in Python with the nibabel library:
import nibabel
mydata = nibabel.load('/path/to/your/mgh/file')
mydata will then be a MGHImage, more information here.
Keep in mind that if you want to manipulate the data vector within this object, you will need to transform it a bit.
Indeed, if you do the following:
raw_data = mydata.get_fdata(dtype="float32")
print(raw_data.shape)
The shape of your "raw" vector will probably look like this: (163842, 1, 1).
Use the squeeze function from numpy to get a (163842,) shape (documentation here).
The reverse operation ((163842,) to a (163842, 1, 1) shape) can be achieved with the atleast_3d function from numpy (documentation here).
This may come handy when you need to create a MGHImage from scratch.
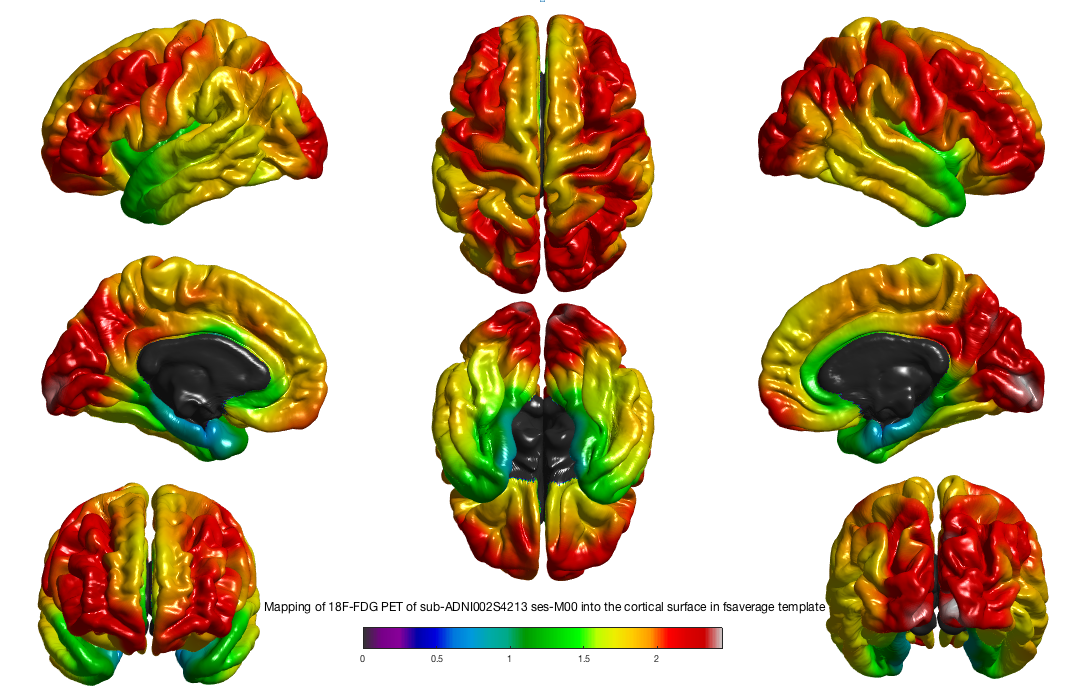
Visualization of the results¶
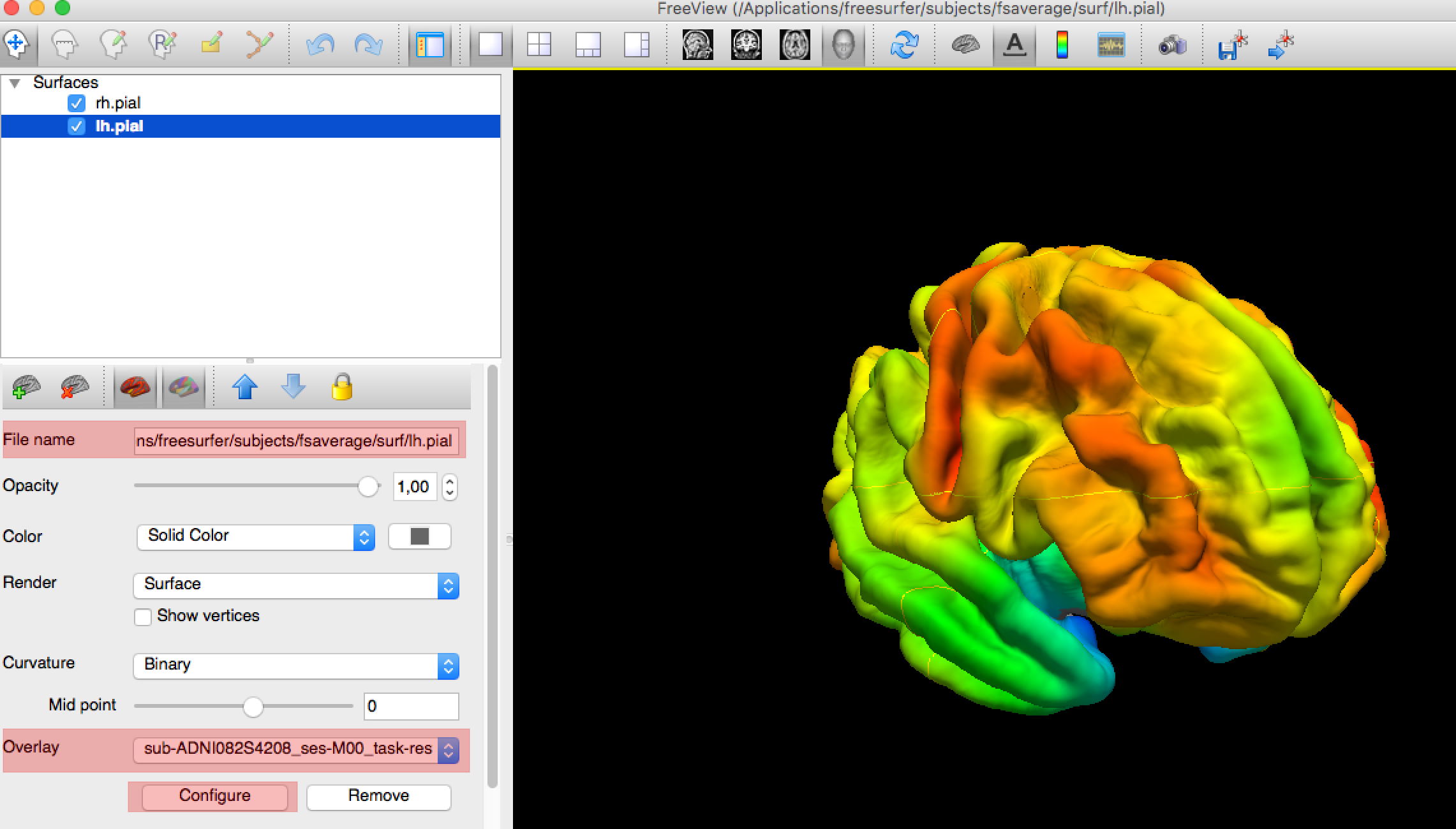
After the execution of the pipeline, you can check the outputs of a subject by running this command (subject moved into FsAverage):
freeview -f $SUBJECTS_DIR/fsaverage/surf/lh.pial:overlay=path/to/your/projected/pet/in/fsaverage/left/hemi \
-f $SUBJECTS_DIR/fsaverage/surf/rh.pial:overlay=path/to/your/projected/pet/in/fsaverage/right/hemi
But you can also visualize your subject cortical projection directly into his native space:
freeview -f path/to/midcortical/surface/left:overlay=path/to/your/projected/pet/in/nativespace/left/hemi \
-f path/to/midcortical/surface/right:overlay=path/to/your/projected/pet/in/nativespace/right/hemi
You will need to adjust the colormap using the Configure button in the left panel, just below the Overlay section.
You can also visualize your surface using the SurfStat tool. Once the SurfStat installation folder is added to your MATLAB path, you can display your surfaces with the following commands:
mydata = SurfStatReadData({'/path/to/left/data', '/path/to/right/data'});
mysurfaces = SurfStatReadSurf({'/path/to/left/surface', '/path/to/right/surface'});
figure, SurfStatViewData(mydata, mysurfaces, 'Title of figure');
You will obtain the following figure:
Contact us !¶
- Check for past answers on Clinica Google Group
- Start a discussion on GitHub
- Report an issue on Github